Join GitHub today
GitHub is home to over 28 million developers working together to host and review code, manage projects, and build software together.
Sign upAre the mass of the generated fragments match the m/zs of fragments ? #13
Comments
This comment has been minimized.
This comment has been minimized.
schymane
commented
Jan 21, 2018
|
Can you please provide us with an example so we can help you interpret your results?
MetFrag does consider the movement of Hs in the assignment of fragments, as these are mobile in the MS/MS due to the charge state. This information (H correction) is included in the results as well so you should be able to follow what is happening.
I will need a peak list and formula/mass and database (or a specific database ID) to help recreate your query and explain further, otherwise you could submit feedback through the MetFragBeta website, but only Christoph will receive those files.
Thanks!
On Sun, Jan 21, 2018 at 6:54 AM +0100, "HC.Ji" <notifications@github.com<mailto:notifications@github.com>> wrote:
Hi,
I tried using the frag.generateFragments function to generate the theoretical fragments of MS/MS. Then I used the get.natural.mass of rcdk package to obtain the mass values of the fragments. But I have a question, should the obtained mass values match the m/z of the MS/MS spectrum? or the mass values should add or reduce a H atom so as to match the m/z of the MS/MS spectrum? Thank you.
Best wishes
—
You are receiving this because you are subscribed to this thread.
Reply to this email directly, view it on GitHub<#13>, or mute the thread<https://github.com/notifications/unsubscribe-auth/AD4a_U6Jmdr1F1PDENxzo53qP37I3tJaks5tMtEGgaJpZM4RltVx>.
|
This comment has been minimized.
This comment has been minimized.
hcji
commented
Jan 21, 2018
|
well, the example codes of I thought the My question is "Can I take the |
This comment has been minimized.
This comment has been minimized.
schymane
commented
Jan 21, 2018
|
Do you have a particular set of MS/MS peaks that you are trying to match? This is what I meant. The answer depends on the context and is easiest to demonstrate with an example.
On Sun, Jan 21, 2018 at 10:14 AM +0100, "HC.Ji" <notifications@github.com<mailto:notifications@github.com>> wrote:
well, the example codes of frag.generateFragments are:
smiles <- "CN(C)CC(C1=C=C(C=C1)OC)C2(CCCCC2)O"
molecule <- parse.smiles(smiles)[[1]]
fragments <- frag.generateFragments(molecule)
I thought the fragments should mean the fragments of MS/MS, right ?
so, I get the mass values of them via:
masses <- sapply(fragments, rcdk::get.natural.mass)
My question is "Can I take the masses as the predicted m/z values of MS/MS ?" or I should add/deduct the mass of "H" from the results ? or some other ?
—
You are receiving this because you commented.
Reply to this email directly, view it on GitHub<#13 (comment)>, or mute the thread<https://github.com/notifications/unsubscribe-auth/AD4a_b9quKNz01Bm_oBKGB4JH2gfVH1wks5tMwAXgaJpZM4RltVx>.
|
This comment has been minimized.
This comment has been minimized.
schymane
commented
Jan 21, 2018
|
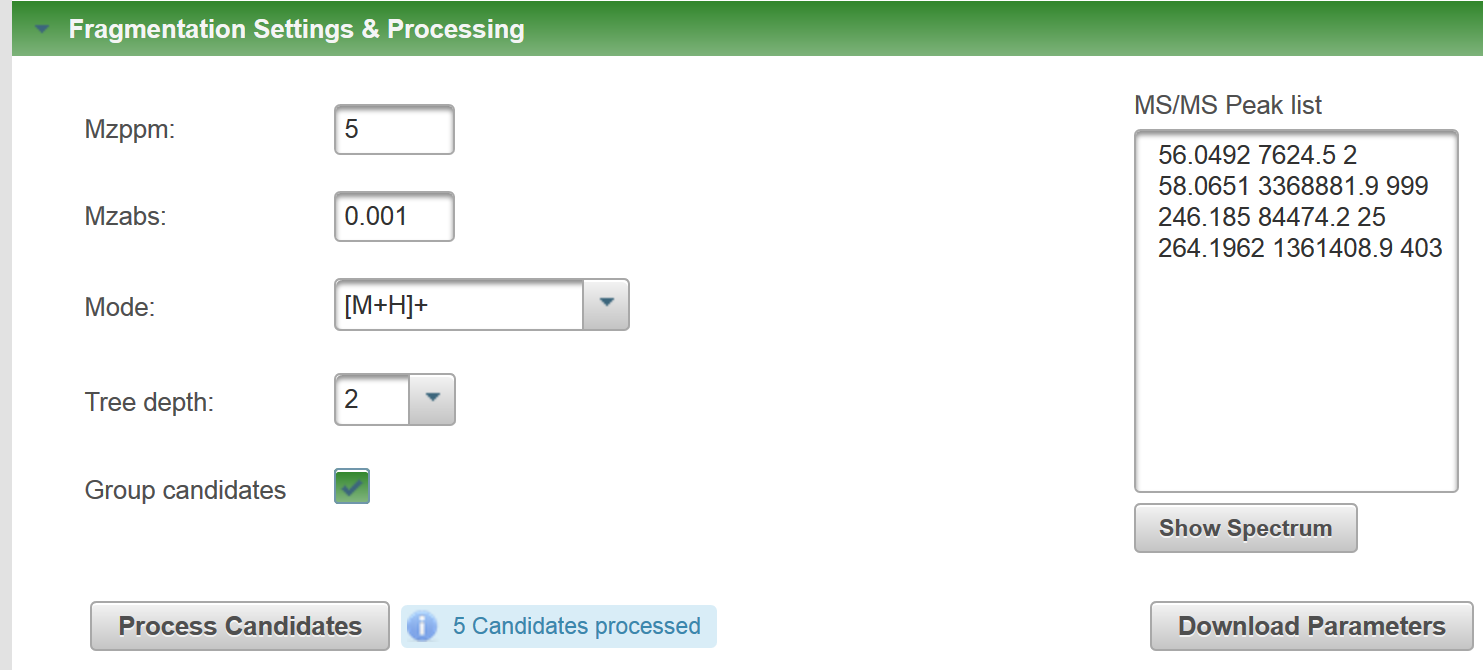
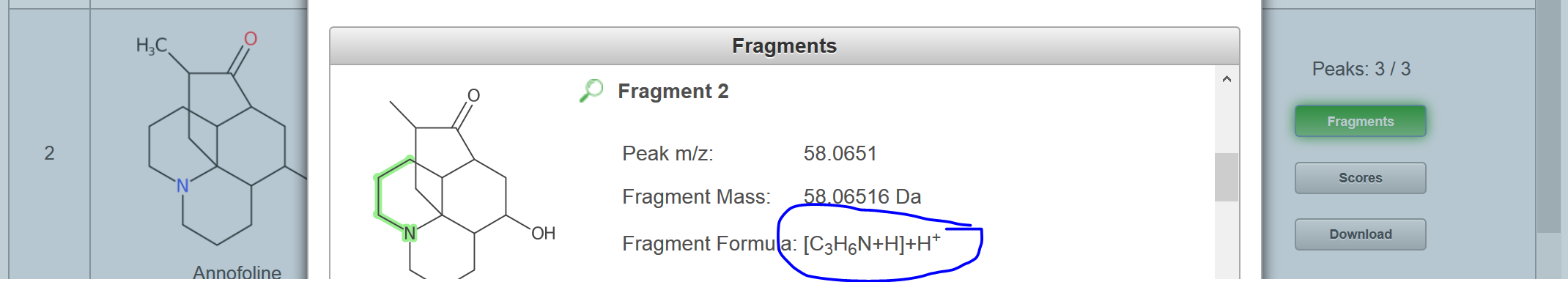
I just ran the formula I generated with your SMILES through MetFragBeta with KEGG and, since Tramadol matched, I took a mass spectrum from Tramadol:
If you look at the fragments generated for the candidates, you will see e.g. for Tramadol, no H adjustment should be necessary (you obtain this picture if you click on "fragments" per candidate: But for the second candidate, there was some adjustment: You may need to try some of your own examples to see how MetFrag deals with this, I hope this help you make a start? |


This comment has been minimized.
This comment has been minimized.
NGAlab
commented
Dec 1, 2018
|
Hi, the previus posts are very interesting and useful, thanks |
This comment has been minimized.
This comment has been minimized.
schymane
commented
Dec 1, 2018
|
Yes you have to match the predicted fragment intensities to the corresponding mass in the experimental (input) spectrum. This information is in the export file somewhere (the fragments explained and corresponding intensities merged together in one column), although not displayed on the web interface explicitly (there you see it graphically).
Thanks for the feedback!
…----------------------------------------------
PI: EnvCheminf @ LCSB
FNR ATTRACT Fellow
emma.schymanski@uni.lu
On Sat, Dec 1, 2018 at 6:46 PM +0100, "NGAlab" <notifications@github.com<mailto:notifications@github.com>> wrote:
Hi,
how can i get the fragment intensities, not only the masses?
the previus posts are very interesting and useful, thanks
—
You are receiving this because you commented.
Reply to this email directly, view it on GitHub<#13 (comment)>, or mute the thread<https://github.com/notifications/unsubscribe-auth/AD4a_WgTvodzomtVnIysdvQWXNzliOurks5u0sCNgaJpZM4RltVx>.
|
This comment has been minimized.
This comment has been minimized.
NGAlab
commented
Dec 1, 2018
|
Thanks for the quick response, smiles <- "CN(C)CC(C1=C=C(C=C1)OC)C2(CCCCC2)O" the masses I can get with the function: how can I get the intensities? Intensities <- ???? Thanks again |
This comment has been minimized.
This comment has been minimized.
schymane
commented
Dec 1, 2018
|
Like I said, these come from the input spectrum using the classic MetFrag approach. MetFrag predicts fragment masses using bond disconnection, but does not predict intensities like CFM-ID. So there is no intensities function ...
…----------------------------------------------
PI: EnvCheminf @ LCSB
FNR ATTRACT Fellow
emma.schymanski@uni.lu
On Sat, Dec 1, 2018 at 7:36 PM +0100, "NGAlab" <notifications@github.com<mailto:notifications@github.com>> wrote:
Thanks for the quick response,
What I would like to know is how I cat get the intensity value for each fragment generated in R with the function written before:
smiles <- "CN(C)CC(C1=C=C(C=C1)OC)C2(CCCCC2)O"
molecule <- parse.smiles(smiles)[[1]]
fragments <- frag.generateFragments(molecule)
the masses I can get with the function:
masses <- sapply(fragments, rcdk::get.natural.mass)
how can I get the intensities?
Intensities <- ????
Thanks again
—
You are receiving this because you commented.
Reply to this email directly, view it on GitHub<#13 (comment)>, or mute the thread<https://github.com/notifications/unsubscribe-auth/AD4a_QT8USfjiflgAse85NHVXGaIUxkJks5u0swrgaJpZM4RltVx>.
|
This comment has been minimized.
This comment has been minimized.
NGAlab
commented
Dec 1, 2018
|
ok, completely clear. thanks! |
hcji commentedJan 21, 2018
Hi,
I tried using the
frag.generateFragmentsfunction to generate the theoretical fragments of MS/MS. Then I used theget.natural.massof rcdk package to obtain the mass values of the fragments. But I have a question, should the obtained mass values match the m/z of the MS/MS spectrum? or the mass values should add or reduce a H atom so as to match the m/z of the MS/MS spectrum? Thank you.Best wishes